
As a regulatory leader, your responsibility doesn’t end with market approval, it begins there. This sentiment echoes across boardrooms and regulatory strategy meetings as post-market surveillance (PMS) becomes a critical pillar of lifecycle management in medical devices. In the times where real-world performance, patient safety, and global compliance are under the spotlight, PMS is a strategic advantage.
According to global regulatory experts, the ability to predict, monitor, and act on post-market data is fast becoming a benchmark for excellence in medical device oversight. While the U.S. FDA and the European Union (EU) both prioritize PMS, their frameworks reflect two distinct philosophies: the FDA’s reactive yet data-driven model versus the EU’s proactive, lifecycle-integrated system under MDR 2017/745.
For companies with global aspirations, understanding these nuances in 2025 is essential to safeguarding brand reputation, achieve compliance, and maintain market access.
Understanding Post-Market Surveillance (PMS)
Post-Market Surveillance refers to the ongoing and systematic collection, analysis, and interpretation of data related to a medical device after it is placed on the market. It helps in identifying risks, evaluating benefit–risk ratios, and supporting regulatory actions such as safety communications, recalls, or labeling updates. PMS is also a fundamental input into the manufacturer’s Corrective and Preventive Action (CAPA) and Quality Management System (QMS) processes.
USA: PMS Requirements Under FDA Regulations
In the United States, PMS activities are primarily governed by 21 CFR Part 803, which covers Medical Device Reporting (MDR).
Key Stakeholders and Responsibilities
- Manufacturers & Importers: Must report device-related deaths, serious injuries, and certain malfunctions.
- User Facilities (e.g., hospitals): Required to report deaths to the FDA and serious injuries to manufacturers (or to the FDA if the manufacturer is unknown).
- Distributors: Not required to report routinely but must maintain records of complaints and provide them upon request.
Timelines for MDR Submission
- 30 calendar days for standard adverse event reports.
- 5 workdays for events requiring remedial action to prevent an unreasonable risk of substantial harm.
- 10 workdays for user facilities to report to the FDA or manufacturers, depending on the nature of the event.
Electronic Medical Device Reporting (eMDR)
Since 2015, all MDR submissions must be made electronically via the FDA’s Electronic Submissions Gateway (ESG). Manufacturers can use:
- eSubmitter software (for MedWatch 3500A XML files), or
- AS2 Gateway using HL7 ICSR-compliant XML formats.
MAUDE Database
Submitted MDRs are made publicly available through the Manufacturer and User Facility Device Experience (MAUDE) database, enhancing transparency and enabling stakeholders to monitor safety profiles.
Recent Developments (2023–2025)
While no major amendments to Part 803 have occurred, the FDA has released:
- Technical updates to the eMDR validation process,
- Guidance encouraging integration of real-world evidence (RWE), and
- Enhancements aligned with IMDRF harmonization goals.
Manufacturers are advised to track the CDRH eMDR enhancements schedule for the latest updates.
EU: PMS Under Regulation (EU) 2017/745 (MDR)
In the EU, PMS is governed by Chapter VII and Annex III of Medical Device Regulation (MDR) 2017/745. The approach is proactive and lifecycle-based, tightly integrated into a manufacturer’s QMS.
PMS Plan and System (Articles 83–84)
Manufacturers must establish and maintain a PMS system, documented through a PMS Plan that outlines:
- Data sources (e.g., complaints, literature, user feedback),
- Analytical methodologies,
- Risk assessment criteria, and
- Corrective action protocols.
PMS Reporting Documentation
- PMS Report: Required for Class I devices and kept internally.
- PSUR (Periodic Safety Update Report):
- Mandatory for Class IIa, IIb, and III devices
- Submitted annually for Class III and Class IIb implantables; every two years for Class IIa and other IIb devices.
- Reviewed by Notified Bodies for Class IIb and III.
Vigilance Reporting (Article 87)
- Serious incidents and field safety corrective actions (FSCAs) must be reported.
- Timelines:
- 2 days for serious public health threats,
- 10 days for death or serious deterioration,
- 15 days for all other serious incidents.
Trend Reporting (Article 88)
Uniquely, EU MDR mandates reporting of statistically significant increases in non-serious incidents or side effects that may impact the device’s benefit–risk profile. Manufacturers must:
- Define trend thresholds in the PMS Plan,
- Regularly analyze data, and
- Report findings if thresholds are exceeded.
EUDAMED Platform
The European Database on Medical Devices (EUDAMED) is a centralized IT system for:
- Device registration,
- Clinical investigations,
- Vigilance reporting,
- PMS activities.
As of 2025, several modules (vigilance, market surveillance, and clinical investigations) are operational, but full mandatory use is still pending a European Commission notice.
Notified Bodies’ Role
For Class IIb and III devices, Notified Bodies review PSURs and upload evaluations into EUDAMED. This adds a layer of external oversight absent in the U.S. system.
USA vs. EU PMS: A Comparative Snapshot
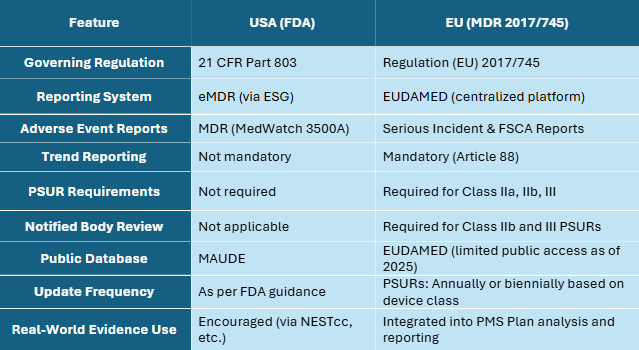
Conclusion
Post-market surveillance is more than a regulatory requirement—it is a cornerstone of ensuring ongoing patient safety, effective risk management, and product performance.
- In the U.S., the FDA’s approach emphasizes reactive adverse event reporting through MDR and public transparency via MAUDE.
- In the EU, MDR has introduced a comprehensive and proactive system, featuring periodic reporting, trend monitoring, and oversight by Notified Bodies.
Manufacturers operating globally must invest in:
- Robust QMS and CAPA systems,
- Integrated regulatory intelligence, and
- Digital tools to handle diverse submission formats and databases (e.g., ESG, EUDAMED).
As 2025 unfolds and regulators continue refining their PMS frameworks, staying informed, agile, and aligned with real-world evidence-driven safety strategies will be critical to success.
About DDReg
DDReg is at the forefront of supporting medical device manufacturers in navigating complex regulatory landscapes like PMS. With over a decade of experience in regulatory strategy, DDReg partners with companies to ensure they not only comply with evolving regulations but also leverage post-market data to enhance safety, streamline reporting, and protect patient well-being. Our expertise in both U.S. FDA and EU regulations allows us to guide you through the intricacies of PMS, offering tailored solutions that drive global compliance and operational success.
Read more from us here: Understanding Software as a Medical Device (SaMD)