
The Most Significant Quality Regulatory Overhaul in 30 Years Is Here
On February 2, 2026, the FDA’s Quality Management System Regulation (QMSR) officially replaced the legacy Quality System Regulation (QSR) under 21 CFR Part 820. This is not an incremental update. It is the most sweeping structural reform to medical device quality regulation since QSR was first enacted in 1996.
At its core, QMSR harmonizes 21 CFR Part 820 with ISO 13485:2016, the globally recognized standard for medical device quality management systems. For manufacturers who already hold ISO 13485 certification, this alignment is a meaningful efficiency gain. For those who do not, the gap between current compliance posture and QMSR requirements may be wider than expected.
Understanding what FDA inspectors will now look for and where manufacturers typically fall short, is no longer optional for quality and regulatory teams.
What FDA QMSR Actually Changes: A Structural Comparison
The architectural difference between QSR and QMSR goes beyond terminology. QSR was prescriptive: it told manufacturers what to do in considerable procedural detail. QMSR adopts a risk-based, outcome-oriented framework aligned with ISO 13485, which shifts regulatory scrutiny from procedure existence to system effectiveness.
Dimension | QSR (21 CFR Part 820, Pre-2026) | QMSR (21 CFR Part 820, Effective Feb 2026) |
Framework Basis | Proprietary FDA requirements | ISO 13485:2016 incorporated by reference |
Approach | Prescriptive | Risk-based and outcome-oriented |
Design Controls | Explicit procedural mandates | Integrated into QMS design and development planning |
Supplier Controls | Defined vendor qualification procedures | Risk-based supplier evaluation per ISO 13485 §7.4 |
CAPA | Separate corrective and preventive action requirements | Unified nonconformity and corrective action process |
Documentation | Document and record control requirements | Explicit document retention periods and traceability requirements |
Inspection Focus | Procedure compliance | QMS effectiveness and risk management integration |
One of the most consequential structural changes is that ISO 13485:2016 is now incorporated by reference into the regulation. This means FDA investigators will use it as a compliance benchmark during inspections, not as guidance, but as enforceable regulatory language.
The New Inspection Regime: What FDA Investigators Are Looking For
The QMSR’s alignment with ISO 13485 changes the inspection dynamic significantly. FDA investigators are trained to assess QMS maturity and systemic effectiveness, not just whether SOPs exist on a shelf.
Key inspection focus areas under FDA QMSR include:
- Risk Management Integration QMSR requires that risk management be embedded across the product lifecycle, not siloed into a single risk file. Investigators will probe whether risk outputs genuinely inform design decisions, supplier qualification, and post-market surveillance, not whether the risk file exists in isolation.
- Design and Development Controls Under ISO 13485 §7.3 (now enforceable under QMSR), design planning, verification, validation, and transfer must be documented with explicit linkages to risk. Gaps in design history files, missing design reviews, or undocumented design transfer criteria are high-probability findings.
- Supplier and Outsourced Process Controls The regulation nowapplies ISO 13485’s §7.4 supplier evaluation framework. FDA inspectors will assess whether supplier risk classification is defensible and whether incoming inspection, audit schedules, and supplier performance monitoring align with the risk profile of purchased goods and services.
- Complaint Handling and Post-Market Surveillance QMSR aligns complaint handling withpost-market surveillance obligations, creating an expectation of closed-loop feedback from the field into design controls and CAPA. Fragmented complaint and vigilance systems are a common target.
- Corrective Action Rigor The unified corrective action process under QMSR requires demonstrated root cause analysis, effectiveness verification, and evidence that corrective actions actually prevent recurrence. Superficial CAPAs that close findings without systemic analysis remain among the FDA’s most frequently cited deficiencies.
Common Deficiencies Manufacturers Should Anticipate
Based on FDA’s historical inspection data under QSR and the documented gaps identified in ISO 13485 third-party audits; the following areas represent the highest-probability findings under the new QMSR inspection regime:
- Inadequate design control documentation, particularly missing or incomplete design outputs, verification/validation protocols, or transfer records
- Risk management files disconnected from QMS processes – risk assessments created as standalone documents rather than integrated inputs to design, manufacturing, and post-market activities
- Supplier qualification records that are incomplete or not risk-stratified, particularly for critical suppliers of components or sterilization services
- CAPA records that close findings without documented effectiveness checks
- Complaint handling processes that fail to consistently trigger MDR or vigilance reporting reviews
- Traceability gaps between design controls, risk management, and production/quality records
- Management review records that lack evidence of data-driven decision-making or resource allocation tied to quality objectives
Strategic Compliance Implications for Manufacturers
The QMSR transition is not simply a documentation exercise. Companies that approach it as such will find themselves exposed during the first post-effective-date inspection cycle.
For ISO 13485-certified manufacturers: The regulatory burden has theoretically decreased, but gap assessments are still necessary. FDA’s interpretation of ISO 13485 requirements may differ from your certification body’s. Unannounced FDA inspections will use the standard as a compliance lens, and auditors trained by notified bodies and FDA investigators will not always interpret requirements identically.
For manufacturers without ISO 13485 certification: The compliance lift is substantial. The entire QMS architecture must be reoriented around the ISO 13485 framework including management responsibility, resource management, product realization, and measurement/analysis/improvement processes. Legacy QSR-era SOPs will not map cleanly to QMSR without deliberate restructuring.
For small and medium-sized manufacturers: Resource constraints make the structural realignment particularly challenging. Prioritizing high-risk product lines and customer-facing quality processes (complaint handling, CAPA, design controls) is a defensible risk-based strategy while broader QMS harmonization is completed.
Actionable Recommendations for Regulatory and Quality Teams
Conduct a formal QMSR gap assessment now. Map your current QMS against ISO 13485:2016 clause by clause, with explicit attention to areas where FDA inspection focus is highest. Do not rely on ISO 13485 certification audit reports alone.
Update your design control procedures and DHF templates. Ensure that design planning, review, verification, validation, transfer, and change records reflect QMSR’s risk-integrated expectations, not QSR-era checklists.
Restructure supplier controls using a documented risk classification system. Assess each critical supplier against quality history, regulatory status, and product risk contribution. Inspection-ready supplier qualification files are non-negotiable.
Strengthen CAPA with effectiveness verification gates. Build effectiveness check timelines and acceptance criteria into CAPA procedures. Train QA teams on structured root cause analysis methods (5-Why, fishbone, fault tree) appropriate to the complexity of the nonconformity.
Integrate post-market surveillance data into management review and design control processes. Create a closed-loop mechanism that brings complaint trends, MDR data, and field failure signals back into the design and risk management system.
Prepare for unannounced inspections. The FDA has historically used unannounced inspection authority for high-risk device categories. QMSR does not change this practice, and a well-maintained quality system should be inspection-ready at any time.
FDA QMSR Implementation Roadmap
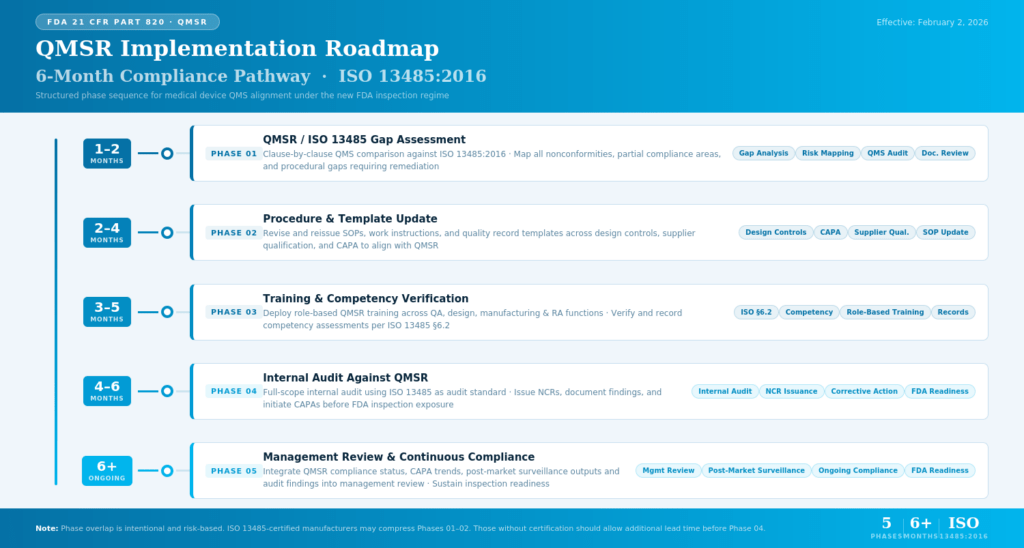
Conclusion
The FDA QMSR marks a fundamental reorientation of how medical device quality is regulated in the United States. By incorporating ISO 13485:2016 as enforceable regulatory language, the FDA has moved decisively away from prescriptive procedural checklists toward an outcomes-based, risk-integrated quality system model, one that rewards genuine QMS maturity and exposes superficial compliance for what it is.
For manufacturers, the message is clear: the transition window has closed, but the compliance work has only begun. A quality system that satisfied QSR inspectors may not withstand the scrutiny of an QMSR-era FDA investigation. The shift demands honest internal assessment, targeted remediation, and a sustained commitment to integrating quality across every function.
The regulatory environment will continue to evolve, but the manufacturers who build durable, risk-based quality systems now will be far better positioned for whatever comes next whether that is a routine FDA inspection, a global market authorization, or a supply chain disruption that tests system resilience. QMSR compliance, done properly, is not a cost of doing business. It is a foundation for doing business well.
Why Choose DDReg for Your QMSR Compliance Journey
DDReg brings specialized expertise across medical device regulatory affairs services, ISO 13485 implementation, and 21 CFR Part 820 compliance, with a track record of supporting manufacturers through complex quality system transitions, FDA inspection preparedness programs, and CAPA remediation across Class I, II, and III device categories. Our consultants have the functional experience to identify where QMS gaps carry the highest inspection risk and the technical precision to close them efficiently, without disrupting ongoing operations.