
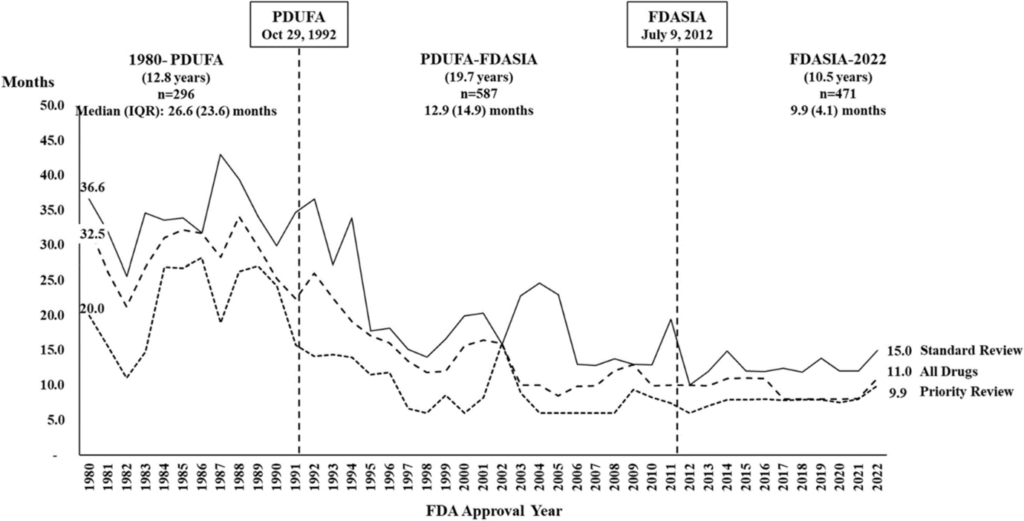
In the past four decades, the number of drugs being approved by the FDA has increased significantly. Interestingly, while the number of products has only increased, the time taken by the FDA to review and eventually market them has steadily decreased. A recent article by Seoane-Vazquez and team shows that while the median time to review a product in 1980 was approximately 26.6 months, it has decreased to 9.9 months in 2022 (Figure 1). One factor that contributed to this was the Food and Drug Administration Safety and Innovation Act (FDASIA) act that allowed the FDA to collect fees from sponsors under the Prescription Drug User Fee Act (PDUFA), that in turn allowed the FDA to gain more resources to be able to fund reviews of biological products, thereby shortening review and approval timelines.
However, another contributing factor to this decrease in review time is regulatory designations. Regulatory designations are assigned by the FDA and, in addition to a shortened review time, also offer benefit to certain products while they are being marketed. Cell and gene therapy products approved in the past five years have benefitted from these regulatory designations, and with their numbers only predicted to rise in the future, it is important to understand what these designations are and how they can help expedite future cell and gene therapy products’ approval processes.
What are the regulatory designations relevant to cell and gene therapy products?
The FDA offers seven regulatory designations that are relevant to cell and gene therapy products. Generally, applications for regulatory designations are filed after the IND is filed, and before the BLA.
Fast Track designation
The Fast Track designation helps to expedite the approval process for those therapies that treat an unmet need in the market for serious diseases and conditions. This includes those products that treat conditions with no other existing treatments, and those products that have an existing treatment in the market but are substantially better in parameters such as safety and efficacy.
The Fast Track designation allows for more frequent meetings and communication with the FDA regarding the development of the drug. It also allows rolling reviews which allow parts of the BLA to be submitted to the FDA as they get completed, instead of waiting for the application to be completed and then submitted. The request for Fast Track designation is made via the sponsor company and the FDA needs to make a decision within 60 days of the request.
Breakthrough Therapy designation
The Breakthrough therapy designation helps to expedite the development and review of those drugs and therapies that treat serious conditions and have proof of substantial improvement over other existing therapies for the disease. Such therapies show improvement over other existing drugs based on a clinically established endpoint such as an established surrogate endpoint or a pharmacodynamic biomarker and generate robust evidence for the same.
A product with the Breakthrough Therapy designation is eligible for all features from the Fast Track designation, in addition to extensive guidance for the development of the product as early as in Phase 1. One of the aims of is to generate robust evidence, so the sponsor should apply for this designation no later than the end of phase 2 meetings. The FDA does not anticipate applications after the BLA has been filed. The FDA must process the application within 60 days of receipt of the application.
Priority Review designation
The Priority Review designation allows for the review of an application to be carried out in 6 months instead of the 10 months under standard review. This designation is granted to a therapy based on whether its expedited review will help improve the treatment of a disease over regular conditions.
The review designation is decided by the FDA, however, the applicant may request for a priority review. The FDA needs to inform the sponsor of the review designation within 60 days of the receipt of the BLA.
Accelerated approval
Accelerated approval helps to expedite the review of a therapy that treats a serious condition, and approval is granted based on data from a surrogate endpoint. It is different from the Breakthrough Therapy designation in that Breakthrough Therapy designation is applied towards improving pre-existing therapies and Accelerated Approval is meant to give access to drugs with data from preliminary stages. The drug company, however, still needs to show the effectiveness of the drug from subsequent studies.
Orphan drug designation
The orphan drug designation is granted to those therapies that treat a rare disease. While deciding on this designation, the FDA considers the mechanism of action of the drug to determine what disease it is intending to treat. If granted, the incentives include up to seven more years of exclusivity in the market, exemption of user fee and tax credits for clinical trials.
Regenerative medicine advanced therapeutic designation (RMAT)
The Regenerative Medicine Advanced Therapeutic (RMAT) designation is assigned to those products that employ the use of cell therapy, therapeutic tissue engineering product, human cell and tissue product, or any combination product (collectively called regenerative tissue therapy). These products should meet an unmet need in the market for a serious disease or life-threatening condition. Gene therapy products generally do not quality for this designation with few exceptions.
The request for RMAT designation should be made by the sponsor company along with the IND submission, or as an amendment to an existing IND. The Office of Tissues and Advanced Therapies (OTAT) informs the sponsor about the decision of the designation no later than 60 days from the receipt of the application.
Rare pediatric disease (RPD) designation
The Rare Pediatric Disease (RPD) designation is used to address drugs or biological products that treat rare diseases in children. They help to bring resources and attention to such products. As an incentive to encourage development in this area, the FDA also launched a Rare Pediatric Voucher (RPV) that, in exchange for development in a rare pediatric product, allowed sponsors to request priority review for a separate product under development. However, this voucher is in the process of discontinuation after 30th September 2024.
How do Cell and Gene therapy products benefit from these designations?
Almost all of the cell and gene therapies approved by the US FDA so far have been assigned at least one of the seven designations above. One reason for this is that these products are generally geared towards diseases that are serious or life threatening and do not have an established treatment. Some examples are depicted below:
Zolgensma
Zolgensma is a CGT product approved by the FDA in 2019 and is used for treatment of Spinal Muscular Atrophy (SMA) in children of age 2 or less. The table below shows the regulatory milestones of Zolgensma. The BLA was filed in November 2018 and, within 6 months in May 2019, Novartis announced that the therapy had been approved. The therapy was designated both Fast Track and Priority Review which likely contributed to the fast approval process.

Kymriah
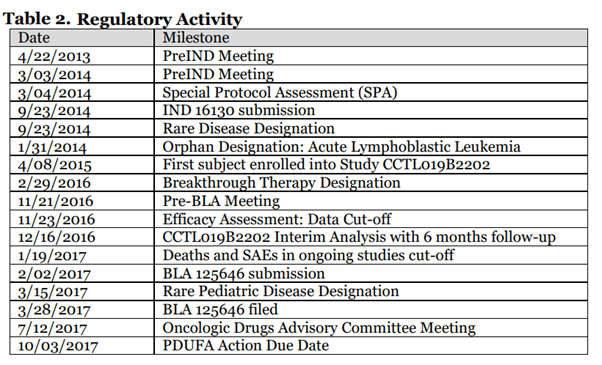
Kymriah is a cell-based gene therapy that uses CAR-T cells to treat lymphoma and leukemia. It was granted many designations, as seen below, including a Special Protocol Assessment (SPA) which allowed sponsors to meet with the FDA and get guidance on clinical trial studies. The BLA was filed in February 2017, and was approved in August 2017.
Overall, regulatory designations can greatly benefit cell and gene therapy products to reach patients quickly and in a safe manner. It is important to understand how these designations work, to fully benefit from the guidance provided by the FDA.