

Introduction
Nitrosamine Drug Substance-Related Impurities (NDSRIs) share structural similarity with API and are therefore unique to every APIs. Drug products containing API that have 2° or 3° amines, when mixed with excipients that have the ability to cause the nitrosation reaction, are responsible for the formation of NDSRIs. There is often a lack of mutagenicity and carcinogenicity study data for NDSRIs from which an acceptable intake (AI) limit can be determined. Indeed, several regulatory challenges arise as a result of uncertainty regarding the presence and acceptability of NDSRI levels in drug products. This leads to applicants conducting unnecessary studies or even discontinuing drug products from the market. Hence, there has been a rise in disruptions of supply and access which can lead to drug shortages and affect patient access to medications. To address these issues, the US FDA recently issued a guidance document that provides risk-based safety assessment for NDSRIs which is applicable to manufacturers and applicants in identifying AI limits for NDSRIs and APIs.
The ‘Recommended Acceptable Intake Limits for Nitrosamine Drug Substance-Related Impurities- Guidance for Industry’ from the FDA provides manufacturers and drug applicants (including both prescription and over the counter (OTC)), a structure for projecting the mutagenicity and carcinogenicity potential of NDSRIs. It provides methodology that uses structural properties of NDSRIs for the determination of AI limits to produce the carcinogenicity data.
This FDA guidance is applicable to: drugs (including both prescription and OTC drug product), prescription and OTC drug products that are in clinical development, biological products (that have fragment and biologic-led combination products which are chemically synthesized).
Background
When FDA was informed about the impurity NDMA i.e., N- nitrosodimethylamine (compound which forms when amines (1°, 2° or 3°) reacts with the nitrous acid) which was found in the Valsartan (an ARB blocker) since then (2018) FDA has been evaluating and managing the impurities of nitrosamine in particular/suspected drug products.
In 2020, FDA introduced the Nitrosamine Guidance, which suggests manufacturers (both API and drug product) to take steps to identify or to reduce the levels of nitrosamine impurities or to use them in our drug product. The Nitrosamine Guidance suggested three step processes for manufacturers and applicants: 1) risk assessment 2) confirmatory testing and 3) reporting of changes implemented to reduce or to prevent the nitrosamine impurities.
As some study suggests that nitrosamine compounds could be potent genotoxic agents in few species of animals, and some are believed to be carcinogenic to humans. So, the APIs and drug product manufacturers are required to take measures to decrease the level of nitrosamine impurities in the drug products.
NDSRIs: Challenges in AI determination
The main challenge in determining the AI limits of NDSRIs is that they are unique to every API and due to this reason, there is limited or no existing safety data. Most NDSRIs do not have a recommended AI limit; the US FDA has provided recommendations for AI limits regarding few NDSRIs. Compounds specific AI limits are determined by using rodent carcinogenic data like TD50 values from published literature. And there are databases such as Carcinogenic Potency Database (CPDB) and Lhasa Carcinogenicity Database (LCDB) from which carcinogenicity data may be obtained. However, many of these studies cannot be relied on for determining AI limits.
In cases where the NDSRI mutagenic potential cannot be characterized adequately, applicants and the US FDA have turned to the structure-activity relationship (SAR) methods to help support in identifying a tested surrogate which has the similar structure and reactivity as the NDSRI, in order to determine an estimation of carcinogenic potency to help identifying a more accurate AI limit. The rationale for choosing the surrogate is critical.
AI limits recommendations based on Predicted Carcinogenic Potency Characterizations (PCPC):
PCPC approach – it explains the techniques that uses PCPC to allocate AI limit to NDSRIs which is based on structural features of NDSRIs whether it is activating or deactivating. If it is activating, then there is an increase in carcinogenic potency and vice versa. This (PCPC) approach integrates the SAR method, which assumes that mutagenic properties of NDSRIs are the result of activation of α-methylation activation. This approach is applicable to those NDSRIs which have C-atoms on both sides of the nitroso group, where the C-atoms are not directly bonded to the heteroatom. Also, this approach does not apply to those NDSRIs (like nitrosated indoles) whose N-nitroso group is located within an aromatic ring. This approach allows manufacturers and applicants to determine the applicable potency category and related AI limits for NDSRIs in API and drug products. If FDA has informed the AI limits directly to manufacturers or applicants or via FDA guidance, then this approach in this guidance should not be applied to NDSRIs.
Specific applications of the PCPC approach – FDA proposed the following five AI limits that are based on the PCPC approach for NDSRIs.
- For potency category 1, the recommended AI limit is 26.5 – means NDSRIs allocated to this category have carcinogenic potency no higher than the class-specific limit for nitrosamine impurities.
- For potency category 2, the recommended AI limit is 100 – means NDSRIs allocated to this category have carcinogenic potency no higher than NDMA & NNK (4-(methylnitrosamino)-1-(3-pyridyl)-1-(butanone)).
- For potency category 3, the recommended AI limit is 400 – means lower carcinogenic potency.
- For potency category 4, the recommended AI limit is 1500 – means low carcinogenic potency.
- For potency category 5, the recommended AI limit is 1500.
PCPC and related AI limits determination –
As per the guidance, the flowchart describes the recommended process of assigning NDSRIs to a predicted carcinogenic potency category, with a corresponding recommended AI limit.
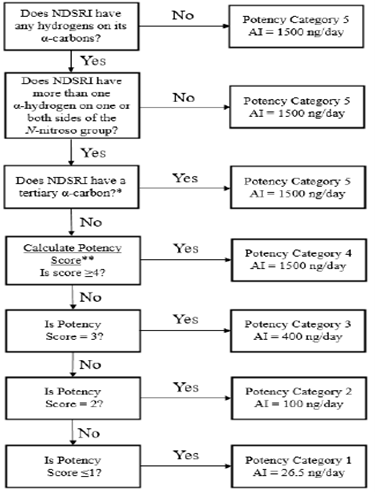
Source: Recommended Acceptable Intake Limits for Nitrosamine Drug Substance-Related Impurities (NDSRIs), US FDA 2023.
Drug product containing multiple nitrosamines: The manufacturer and applicant are required to contact the agency if multiple nitrosamine impurities and/or small molecule nitrosamine are determined, and the entire nitrosamines level surpasses the AI limits for the most potent nitrosamine in the drug product.
NDSRIs AI limits: Risk of formation in certain APIs
FDA has determined that certain APIs having 2° or 3° amines are at risk of forming NDSRIs because certain excipients contain residual nitrites, and the manufacturing process may lead to formation of the NDSRIs. Based on chemical structure analysis, these AI limits based on predicted carcinogenic potency need to be applied to the NDSRIs when an active ingredient contains the hypothetical nitrosated form of 2° amines and dimethyl 3° amine groups.
Conclusion:
Certain APIs having 2° or 3° amine are more prone to undergo nitrosation reaction and forming NDSRIs, when they are formulating with excipients containing residual nitrites. If possible, manufacturers should try to avoid the addition of such excipients or try to mitigate the level of the same. These impurities are carcinogenic in nature when exposed to long term. More studies need to be performed to determine the relation between the amount/level of NDSRIs and carcinogenicity.
References and Further Reading:
- US FDA. Recommended Acceptable Intake Limits for Nitrosamine Drug Substance-Related Impurities (NDSRIs)- Guidance for Industry. United States Food and Drug Administration. 2023