
The rules governing the clinical trials in the UK have not seen a change of this scale since 2004. With the UK Clinical Trials Amendment Regulations 2025 coming into force, sponsors need to take immediate steps to ensure compliance before April 2026.
These amendments are not simply administrative paperwork – they impact clinical trial design, safety monitoring, data management, and regulatory reporting. Lack of compliance can trigger approval of postponements, regulatory challenges, or trial suspension.
We are breaking down the key changes, outlining sponsor responsibilities, and providing actionable steps to help organizations maintain a competitive edge.
Why the UK Clinical Trials Amendment Regulations 2025 Matter?
The UK’s new Clinical Trials Regulation 2025 marks a modernized shift to renew UK trial activity and reinforce its position in global research. The MHRA aims to create a framework that prioritizes efficiency, balance, and transparency. Here’s why these amendments are made to reshape the landscape:
Increasing Global Competitiveness
UK-based trials have declined in recent years. This simplifies approvals and positions in the UK to compete more effectively with other regions’ clinical trials. These new rules release a 30-day statutory review timeline for the initial submission process.
Example- A sponsor planning a gene therapy trial can now rely on defined timelines, avoiding delays that previously slowed participants’ recruitment.
Risk-balance Oversight
The Notification Scheme allows regulators to focus on high-risk, innovative therapies such as gene and cell therapies, ensuring resources are allocated where they matter most. This is now legally recognized, removes the unnecessary regulatory framework for trials involving low risk, and approved medicines.
Transparency
Registration of trials and sharing of results are now legally required. This ensures that all trial data- including studies that fail- contribute to scientific knowledge, reduce research waste, strengthen public confidence, and gain trust.
Modernized Standards
By mandating the ICH E6(R3) GCP Guidelines, the UK has become one of the first leading regions for implementing modern, data-driven clinical trials. This enables decentralized and digital trials.
Evolution Process of UK CTR
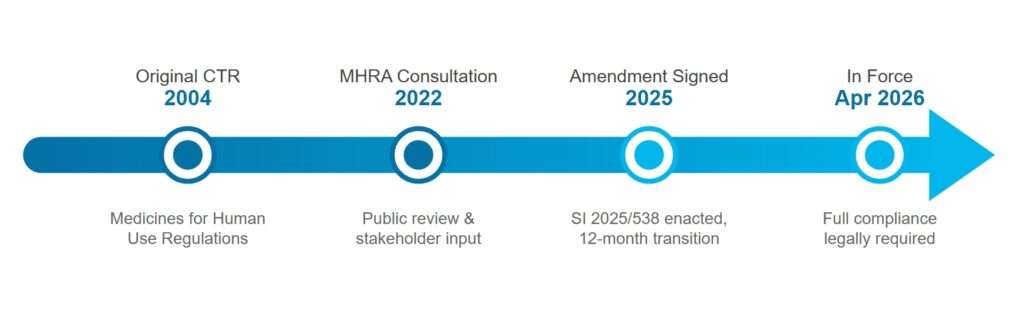
Understanding UK Clinical Trial Compliance Requirements
From 28 April 2026, following the UK Clinical Trials Amendment Regulations 2025 is a legal requirement, not an option. Sponsors must have their operational frameworks fully prepared, as non-compliance now carries the risk of criminal penalties and violation notices issued by the MHRA.
Legal Compliance Includes:
Transparency and Disclosure- The 90 days/12months rule
- Trial Registration: All trials must be registered in a WHO- compliant registry before enrolling the first participant or within 90 days of approval, whichever is sooner.
- Result Reporting: A summary of trial results must be published within 12 months of completion.
- Summaries: Sponsors are required to provide participants with clear, non-technical information and participant engagement.
- Recruitment Commencement: All approved trials are expected to recruit their first UK participant within 2 years of approval. If no recruitment occurs within this window and an extension has not been granted, the approval will lapse.
Follow Good Clinical Practice (GCP)
- Includes ICH E6 (R3): UK-based clinical trials must align with the latest international GCP standards, following quality-by-design along with monitoring through a risk-based approach.
Participant focused Documentation
- Diversity Plans: Applications must include an Inclusion and Diversity Plan, with clear, data-driven justification for recruitment strategies
- Terminology Updates: All contracts and protocols must replace “subject” with “participant”, “amendment” with “modification”, and “trial site” with “trial location”, reflecting a more patient-focused approach.
Easy Application and Modification Pathways
- Combined Review: Sponsors must use the Integrated Research Application System (IRAS) to facilitate a single, parallel review by both the MHRA and the Ethics committee.
- 3 Modifications tiers (1) Substantial Modifications- Major changes requiring full review (2) Minor Modifications- Internal Record-Keeping required (3) Changes in important details- Only Notification
Records and Data Store
- Current Clinical Trials: These trials submitted before this deadline can retain TMFs for 5years, but medical records must still follow the 25-year rule.
- Record Retention for 25-years: The trial Master File (TMF) and medical records for trials submitted on or after 28 April 2026 must be retained for 25 years.
Main Changes in UK CTR Amendment (April 2026)
Here are some common features that will be modified as per the UK CTR Amendment in April 2026, and a comparison with the current process for more clarity.
Features | Current Process (2004) | 2025 Amendments (Post-April 2026) |
Submission Type | Hybrid | Mandatory Combined Review |
Timeline for review | Legally 30 days or extend | Strictly 30-days for initial decision |
Low-Risk Timeline | Standards authorization required | Notification scheme- 14-day review window for eligible substantial modifications |
Changes | Substantial Vs Non-Substantial | Tiered system |
GCP | ICH E6 (R2) | ICH E6 (R3)- Quality by design |
Transparency | Guidance-based registration & reporting | Registration within 90 days & results within 12 months |
Terms change | Subjects | Participants |
Records Keeping | 5 years | 25 years |
Patient Requirements | Recommended | Legally Required |
What are Sponsors' Responsibilities Under the 2025 Regulations
Sponsors play a main role in ensuring that each aspect of a clinical trial meets the updated UK requirements. Primary responsibilities of Sponsors include:
- Documentation Inspection – Make sure protocols, investigator brochures (IB), consent forms, and trial master files (TMF) are correct and up to date.
- Data Protection – Ensure that clinical trial data is securely stored, fully auditable, and respecting privacy of regulations
- Staff Training and Reports – Train all personnel on updated processes, digital submission systems, and reporting requirements.
- Safe Reporting within time – Report serious adverse events (SAEs) and unexpected adverse events within the new timelines.
- Internal Audits for gap analysis – Conduct regular audits to find compliance gaps before external audits
For the Smooth Implementation prepare correctly; to avoid last-minute difficulties, sponsors should start preparations immediately without delays:
- Conduct internal gap analyses within the company.
- To clear and reduce errors, regularly update the Standard Operating Procedures (SOPs)
- Staff training and knowledge tests on the latest regulatory changes
- Get guidance from experts
- Consultation from clinical regulatory professionals
- Test your digital systems to ensure readiness
Conclusion
The UK Clinical Trials (Amendment) Regulations 2025 were developed as an implacable law from advisory guidance. In April 2026, Stricter transparency rules, 25-year record maintenance, mandatory diversity plans, and alignment according to ICH E6(R3) efficacy guideline, now compliance is not an option, it must be part of every trial from the starting phase. With expanded enforcement law, the consequences of not following include regulatory penalties and an effect on reputation.
Sponsors who prepare early won’t just avoid disruption; they’ll gain a competitive edge in a faster, more accountable UK clinical research environment.
Why Choose DDReg for Clinical Regulatory Services?
DDReg offers strategic, clinical regulatory services support designed for fast approvals while ensuring full compliance with new global requirements. Our professional team blends complex regulatory expertise with structured processes to maintain regulatory submissions. We provide precise, risk-based guidance that ensures the start-up, simplified regulatory approach, and consistent compliance across markets. This approach increases the chances of success in international markets.