
Structured Product Labeling (SPL) was first introduced in 2006 by the US Food and Drug Administration (FDA) with the aim to enhance the communication of product, quality, and facility information to label submissions. The move to the electronic Extensible Markup Language (XML) format, compared to the PDF format, has further streamlined drug label review processes, and is associated with other benefits such as enhanced information exchange between computer systems, more efficient exchange of changes in labels between manufacturers and the US FDA, automation in comparing drug data information, and more [1]. Indeed, it is essential to transform the narrative material found in the SPL documents into computer-readable data in order to effectively distribute drug safety information to keep in line with innovation and emerging technologies that are being used in the industry.
General structure of the SPL
The general structure of the SPL is as follows [2]:
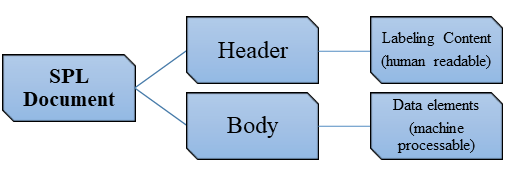
The labeling content constitutes elements such as the text which is human readable. The machine processable elements includes product ingredients, administration route, the dosage form, the product appearance, information on quantity etc.
Stylesheets are utilized to display the labeling content that is stored in XML. A stylesheet is a file that contains formatting guidelines for XML documents. One XML file can be used with many stylesheets to generate various output formats and documents. For instance, the SPL content can be modified to create a USPI as well as advertising materials, product websites, and package components using a single SPL XML source file and numerous stylesheets.
Three e-Files for Registration & Listing – SPL Format [3]
- NDC Labeler Code Request
- Establishment Registration
- Content of labeling (CoL)/Listing
Challenges in SPL
Though the SPL is a small part of the electronic common technical document (eCTD), it is associated with many challenges that can hinder the compilation of the SPL and lead to delays in submissions. Pharmaceutical product labeling is subjected to stringent regulations, and it is crucial to maintain compliance with region-specific labeling requirements. Understanding the technical know-how of the XML is imperative or else organizations can face technical issues that could lead to SPL rejection.
Furthermore, it is imperative to stay updated with the ever-changing regulations especially as they become more stringent. Lack of knowledge on the most updated regulatory landscape could result in multiple re-working when compiling the SPL. This would, in turn, lead to delays and even rejections by the US FDA. So, it is important for companies to be aware of the most recent updates in regulations with relevance to SPL submission.
Finally, a key aspect that may be overlooked, is making sure that all tools used in compiling the SPL are compliant with the US FDA requirements such as the Title 21 CFR Part 11 [4]. Non-compliance or any technological deficiencies may result in slowing down the SPL submission process.
SPL capabilities at DDReg
The team at DDReg is highly experienced in drafting and developing package inserts and medication guides for US FDA submission. Additionally, the team is qualified and extensively trained in XML and converting PDF versions to XML-format SPL files in line with the US FDA requirements. The team is dedicated to providing support in converting product labels to validated SPLs as well as version-controlled procedures to manage the SPL lifecycle for pharmaceutical and allied organizations.
References and additional material:
[1] US FDA. Guidance for Industry: Providing Regulatory Submissions in Electronic Format- Content of Labeling. United States Food and Drug Administration. April 2005.
[2] US FDA. SPL Implementation Guide for FDA Content of Labeling Submissions. Release 1. United States Food and Drug Administration. December 2004.
[3] US FDA. SPL 101 “The Basics”. United States Food and Drugs Administration. 2022
[4] Code of Federal Regulations. Title 21 CFR Part 11: Electronic Records; Electronic Signatures. 2022